


Visit Our Sponsor
Westlab Pharmacy

Westlab Pharmacy
4401 W Newberry Rd #A5
Gainesville, FL 32607-2290
352-371-8111
800-4WESTLA

Lower Motor Neuron Disorders

Introduction:
 One of the causes
of acute quadriparesis or quadriplegia is that of diffuse lower motor neuronal
(LMN) dysfunction, resulting in diminished to absent reflexes in all 4
legs. In this instance the immediate differential diagnosis must include
acute polyradiculoneuropathy (previously discussed), Coral snake envenomation,
tick paralysis, and botulism. Other causes which normally result in normal
reflexes with progressive weakness on exercise include polymyositis, myasthenia
gravis, and sub-acute organophosphate (OP) intoxication. The nature of
these diseases are that they affect some portion of the lower motor unit.
Polyradiculoneuropathy affects the myelinated fibers of the motor nerves.
Coral snake envenomation, tick paralysis and botulism result from toxins
which alter the release of acetylcholine at the neuromuscular junction.
Myasthenia gravis and sub-acute OP intoxication alter the release or responsiveness
of the neuromuscular junction to acetylcholine on repeated neural activity.
Polymyositis directly affects the muscle. The former conditions result
in flaccid paralysis, while the latter ones result in weakness, stiffness
and collapse on exercise. Coral snakes are limited in their distribution
and it should be considered in cases of LMN disease in areas where they
are endemic. The ticks which are associated with tick paralysis vary and
are present throughout the world. Tick paralysis appears to be seasonal,
but can occur at any time. Botulism can occur from food poisoning or from
alterations in the intestinal flora. Dogs are fairly resistant to botulinum
toxin, but intoxication is occasionally seen.
One of the causes
of acute quadriparesis or quadriplegia is that of diffuse lower motor neuronal
(LMN) dysfunction, resulting in diminished to absent reflexes in all 4
legs. In this instance the immediate differential diagnosis must include
acute polyradiculoneuropathy (previously discussed), Coral snake envenomation,
tick paralysis, and botulism. Other causes which normally result in normal
reflexes with progressive weakness on exercise include polymyositis, myasthenia
gravis, and sub-acute organophosphate (OP) intoxication. The nature of
these diseases are that they affect some portion of the lower motor unit.
Polyradiculoneuropathy affects the myelinated fibers of the motor nerves.
Coral snake envenomation, tick paralysis and botulism result from toxins
which alter the release of acetylcholine at the neuromuscular junction.
Myasthenia gravis and sub-acute OP intoxication alter the release or responsiveness
of the neuromuscular junction to acetylcholine on repeated neural activity.
Polymyositis directly affects the muscle. The former conditions result
in flaccid paralysis, while the latter ones result in weakness, stiffness
and collapse on exercise. Coral snakes are limited in their distribution
and it should be considered in cases of LMN disease in areas where they
are endemic. The ticks which are associated with tick paralysis vary and
are present throughout the world. Tick paralysis appears to be seasonal,
but can occur at any time. Botulism can occur from food poisoning or from
alterations in the intestinal flora. Dogs are fairly resistant to botulinum
toxin, but intoxication is occasionally seen.
Diagnostic Approach:
 The diagnosis of motor
unit disease requires looking for systemic illness, ruling out obvious
vertebral disease and identifying the specific changes of the disease.
This includes many of the test employed in looking for cervical and TL
disease, including CSF tap and analysis. On the other hand, serum muscle
enzyme levels (CPK, AST, LDH and aldolase concentrations) should be run.
In some cases, pre- and post-exercise levels of CPK and lactate can help
confirm the diagnosis. In addition, serum cholinesterase levels are needed
to evaluate the potential for OP intoxication. The most important aspect
of the examination are EMG studies, including needle EMG, nerve conduction
velocities and repetitive nerve stimulation. Muscle and/or nerve biopsy
may allow histopathologic diagnosis, muscle immunohistology, fiber typing
and enzyme measurements. In cases of suspected myasthenia gravis, response
to edrophonium HCl, a short acting anticholinergic drug, can be used. Edrophonium
given intravenously at 0.1-0.2 mg/kg can produce dramatic improvements
in muscle strength in 1-2 minutes in patients in myasthenic crisis or who
is exercise intolerant. On the other hand, it can worsen the signs of OP
intoxication. For this reason, I prefer to perform edrophonium testing
under anesthesia during the EMG procedure.
The diagnosis of motor
unit disease requires looking for systemic illness, ruling out obvious
vertebral disease and identifying the specific changes of the disease.
This includes many of the test employed in looking for cervical and TL
disease, including CSF tap and analysis. On the other hand, serum muscle
enzyme levels (CPK, AST, LDH and aldolase concentrations) should be run.
In some cases, pre- and post-exercise levels of CPK and lactate can help
confirm the diagnosis. In addition, serum cholinesterase levels are needed
to evaluate the potential for OP intoxication. The most important aspect
of the examination are EMG studies, including needle EMG, nerve conduction
velocities and repetitive nerve stimulation. Muscle and/or nerve biopsy
may allow histopathologic diagnosis, muscle immunohistology, fiber typing
and enzyme measurements. In cases of suspected myasthenia gravis, response
to edrophonium HCl, a short acting anticholinergic drug, can be used. Edrophonium
given intravenously at 0.1-0.2 mg/kg can produce dramatic improvements
in muscle strength in 1-2 minutes in patients in myasthenic crisis or who
is exercise intolerant. On the other hand, it can worsen the signs of OP
intoxication. For this reason, I prefer to perform edrophonium testing
under anesthesia during the EMG procedure.
Specific Disorders
Polymyositis:
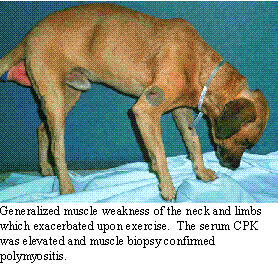 Idiopathic polymyositis
is a diffuse inflammation of the skeletal muscles. An autoimmune process
is suspected, in most cases. Hypergammaglobulinemia, positive titers for
serum antinuclear antibody (ANA), and elevations of circulating anti-muscle
antibodies (also demonstrable by indirect immunohistology) have been found
in many cases. There is generalized weakness which worsens on exercise.
These patients also have a stiff stilted gait, lameness and pain on muscle
palpation. Occasionally, generalized loss of skeletal muscle mass is evident.
Megaesophagus may be present with regurgitation and aspiration pneumonia.
Idiopathic polymyositis
is a diffuse inflammation of the skeletal muscles. An autoimmune process
is suspected, in most cases. Hypergammaglobulinemia, positive titers for
serum antinuclear antibody (ANA), and elevations of circulating anti-muscle
antibodies (also demonstrable by indirect immunohistology) have been found
in many cases. There is generalized weakness which worsens on exercise.
These patients also have a stiff stilted gait, lameness and pain on muscle
palpation. Occasionally, generalized loss of skeletal muscle mass is evident.
Megaesophagus may be present with regurgitation and aspiration pneumonia.
The main features on examination are the stiff, stilted gait. Neurologic examination (in a quiet room) is often within normal limits, except for possible muscle pain on palpation. Other signs relate to the presence of aspiration pneumonia, including fever and listlessness. During the acute phase, muscle enzymes will be elevated. The diagnosis is supported by the findings of an abnormal needle EMG with normal nerve conduction velocities. There is occasionally a decremental response on repetitive nerve stimulation. Muscle biopsy will confirm the diagnosis.
 The treatment
of polymyositis is with immunosuppressive doses of corticosteroids. Cases
with aspiration pneumonia have a poor prognosis, since treatment with immunosuppressive
drugs cannot be initiated. In those cases where corticosteroids are effective,
monitoring the CPK may be useful in determining the course of therapy.
If the CPK decline to normal for 1 month, the steroid medication can be
stopped. The prognosis is generally good if esophageal, pharyngeal and
laryngeal muscles are not severely affected. The megaesophagus may resolve
with therapy or be permanent.
The treatment
of polymyositis is with immunosuppressive doses of corticosteroids. Cases
with aspiration pneumonia have a poor prognosis, since treatment with immunosuppressive
drugs cannot be initiated. In those cases where corticosteroids are effective,
monitoring the CPK may be useful in determining the course of therapy.
If the CPK decline to normal for 1 month, the steroid medication can be
stopped. The prognosis is generally good if esophageal, pharyngeal and
laryngeal muscles are not severely affected. The megaesophagus may resolve
with therapy or be permanent.
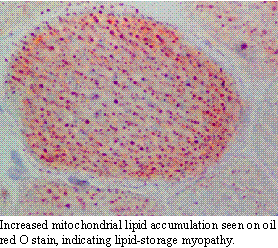
Recently, (probably due to a more aggressive approach to muscle disease
including biopsy and special staining procedures) many additional muscle
diseases which present similarly to polymyositis have been uncovered. Lipid-storage
myopathy is one of these. These patients present with stiff, stilted painful
gaits with tremendous pain on muscle palpation. Histopathology reveals
increases in mitochondrial lipids on special (oil red O) stains. These
patients may respond dramatically to coenzyme Q-10 and l-carnitine supplementation.
Myasthenia gravis:
Myasthenia gravis has been considered a rare disease; however, the adult-onset form seems to be on the rise. Whether this represents improvements in diagnostic criteria and methodology or a real increase in the condition remains unsure. On the other hand, this may represent an increase, similar to that for other immune-related diseases. It has been reported in young dogs (juvenile form) and adult dogs and cats. The juvenile form represents a genetic problem where there are too few acetylcholine receptors at the neuromuscular junction. The adult form represents an auto-immune disease where there is the production of antibodies against the acetylcholine receptor, demonstrated by elevations in circulating anti-receptor antibodies. With either the juvenile form or the adult form, myasthenia represent a functional deficiency of sustainable release of acetylcholine from the pre-synaptic neuron at the neuromuscular junction. This distinguishes myasthenia from the myasthenic-like disease of OP intoxication.
The disease can be seen in any adult dog or cat, the young adult German Shepherd dog being overly represented. There may be an inciting febrile episode, but often no inciting cause is found. Animals present with weakness and exercise intolerance. There may be changes in bark, difficulty in swallowing and prehending food. Regurgitation and megaesophagus may be present. Some cases present in a myasthenic crises where the animal has collapsed and is unable to rise. At this time, there may be muscle flaccidity.
In uncomplicated myasthenia gravis (those without pulmonary pathology
from aspiration pneumonia), the animal demonstrates progressive weakness
on movement which improves with rest. Mild cases may be difficult to differentiate
from animals with polymyositis. Except when in a myasthenic crisis, neurologic
assessment is usually normal. The diagnosis is suspected by demonstration
of a normal needle EMG, normal nerve conduction velocities and decremental
response to repetitive nerve stimulation. (We normally stimulate at 10/second,
recording the first 9 stimulations. An abnormal response is when the 4th
and 9th waves are <85% of the initial wave) Confirmation
of the diagnosis is made by demonstrating elevated serum anti-acetylcholine
receptor antibodies (>0.06 ng/ml). Additional confirmation can be made
by positive response to edrophonium HCl either in the conscious animal
or during the EMG procedure. (Remember the OP intoxication can look like
myasthenia. I, therefore, do not give edrophonium until I have checked
serum cholinesterase levels.)
The treatment of myasthenia is to suppress the production of antibodies
and to increase acetylcholinergic function. The former is usually done
by the administration of prednisolone at 1-2 mg/kg/day. The dose is reduced
while monitoring the serum anti-receptor antibody levels. The latter is
done by giving pyridostigmine at 10-60 mg twice a day (depending upon the
size of the patient). During a myasthenic crisis, intramuscular injections
of neostigmine (0.5-2.5 mg) may be given in cats and dogs. The prognosis
is guarded. In uncomplicated cases, the signs may resolve and go into remission.
In cases with aspiration pneumonia, the prognosis is usually poor. Recently,
immunotherapy has offered new hope in treating refractory cases. By inoculation
of patients with purified anti-receptor antibodies, they produce anti-idiotypic
antibodies which can help control their altered immunity. Since this may
be a disorder which is developed in response to environmental intoxication
with resultant failure of the healing process, dietary supplementation
and antioxidant support may help prevent this disease.
Organophosphate Intoxication:
Acute, subacute and chronic forms of organophosphate intoxication occur. The chronic form is due to a dying-back neuropathy which occurs relatively infrequently and was caused by older forms of organophosphates no longer in use. Treatment of the acute and subacute forms of organophosphate intoxication will be discussed here.
In the acute syndrome, the main signs are associated with muscarinic receptor over stimulation, including salivation, vomition, diarrhea and tachycardia. Central cholinergic receptor over stimulation results in hyperexcitability, body tremors and seizure activity. The muscarinic signs are best treated with atropine (0.04 mg/kg IV or SQ), which blocks the muscarinic receptor overstimulation. The central cholinergic signs may be treated with diazepam (0.5-1.0 mg/kg IV), if necessary to stop seizure discharges. The seizures can be controlled further with pentobarbital (1 mg/kg IV) and/or phenobarbital (1-2 mg/kg IV). Additional therapy for acute organophosphate intoxication (in the dog, only), within the first 24 hours, should include the use of pralidoxime chloride (2-PAM, 20 mg/kg IV, IM or SQ) in order to bind any free organophosphate in the blood stream and aid in the elimination of the intoxicant. Following the initial 24 hours, the organophosphate compound will already be irreversibly bound to acetylcholinesterase and 2-PAM may not be beneficial. To maintain the control over the muscarinic receptor overstimulation and to prevent the development of signs of subacute organophosphate intoxication, continued treatment of acute organophosphate intoxication should include diphenhydramine (Benadryl) orally (1-4 mg/kg) every 8 hours for, at least, 21 days of total therapy. Continued use of atropine in combination with diphenhydramine is not recommended, since overt sedation can result. One initial dose of atropine followed by diphenhydramine for maintenance is sufficient to minimize the signs of acute organophosphate intoxication.
The clinical signs of subacute organophosphate intoxication include muscle weakness, exercise intolerance, bradycardia, central depression, acute dystonia and central vestibular signs and are secondary to diminished nicotinic and central cholinergic receptor function. Since the muscarinic receptor rapidly develops tolerance, the muscarinic signs of acute organophosphate intoxication decrease, and the signs of nicotinic receptor paralysis dominate. Nicotinic receptor paralysis may be delayed in development for 7-10 days after exposure and are much more likely to occur following high or repeated exposure to organophosphate. Anti-muscarinic drugs and 2-PAM are ineffective in treating these signs. Diphenhydramine will stabilize the nicotinic receptor and improve the clinical signs. In subacute organophosphate intoxication, nicotinic paralysis often results in a decremental response seen following repetitive nerve stimulation, producing a myasthenic-like syndrome. This decremental response is worsened by edrophonium HCl, but resolves following diphenhydramine therapy. The mental depression caused by central cholinergic receptor dysfunction and acute dystonia improve with diphenhydramine treatment, as well. Treatment with diphenhydramine should be started IV or IM medication (IM only in the cat) at 4 mg/kg every 4-6 hours. Parenteral therapy should continue until there is improvement in clinical signs, usually by 24-72 hours. Then, oral diphenhydramine is continued at 4 mg/kg every 8 hours. In small and large dogs, sedation may occur at this dosage. Reducing the dose of diphenhydramine to 1-2 mg/kg may be necessary in these patients. Oral diphenhydramine should be continued for, at least, 3 weeks, or until the acetylcholinesterase enzyme has been replaced. While it may require a number of weeks for the plasma cholinesterase to return to normal levels, the functional pool (approximately 20% of the total) usually returns within 3 weeks. This method of therapy for the subacute organophosphate disorders has been highly successful except where the intoxication is extreme. While the exact mechanism of action of diphenhydramine is not known, it is an antihistamine with anticholinergic properties. It is useful as a central anticholinergic, reducing cholinergic activation. While it does not block nicotinic receptor activity, diphenhydramine appears to prevent nicotinic receptor overstimulation. Over the past 10 years, diphenhydramine has been used successfully to reduce the morbidity and mortality of subacute organophosphate intoxication in the dog and cat.